- 微生物
- DNA
- 转录与调控
- 单细胞空间组
- 蛋白与代谢
- 常规测序


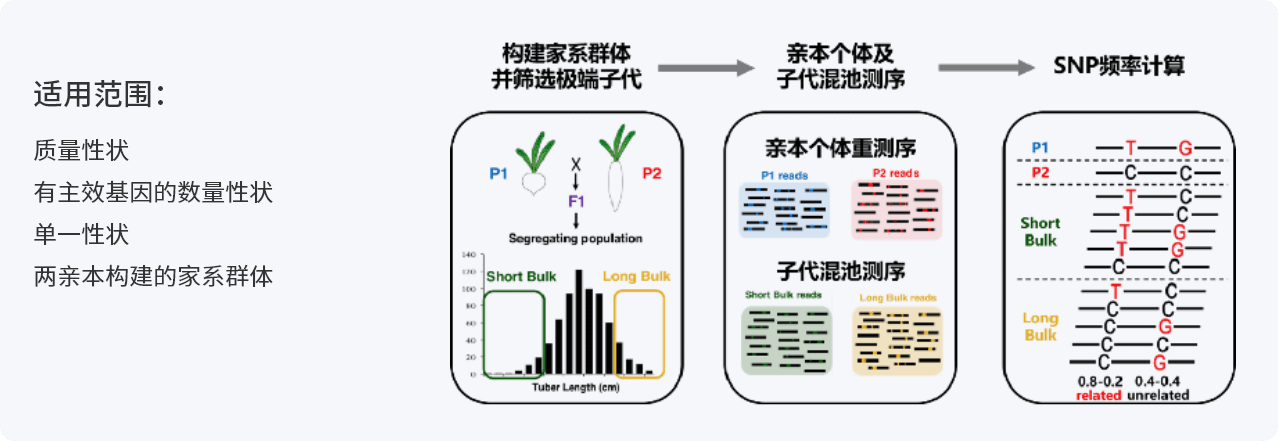
BSA(Bulked Segregant Analysis):集群分离分析法,是指利用目标性状存在差异的两个亲本构建家系,在子代分离群体中,选取目标性状个体构建DNA混合池,结合高通量测序技术对混合DNA样本测序,根据基因型频率的差异筛选基因组上与目标性状相关联的位点,并对其进行功能注释,进而研究控制目标性状的基因及其分子机制。
产品类型

- 优势1
- 应用场景更广阔
- 美吉针对不同材料类型(缺失1亲本;缺失两亲本;子代单混池等)的BSA基因定位分析分析有不同的分析策略和生信分析流程
- 优势2
- 交付内容更丰富
- 美吉的交付内容包含3种主流的SNP频率计算方法的结果,更全面更具体的定位候选区域,且美吉还交付定位区域内的引物设计结果便于客户后续位点验证
- 优势3
- 售前售后更周到
- 美吉拥有专业的售前咨询和细致的售后服务,可为客户的不同分析需求提供更专业的指导,响应速度快,质量佳,可为客户服务到文章发表

测序流程
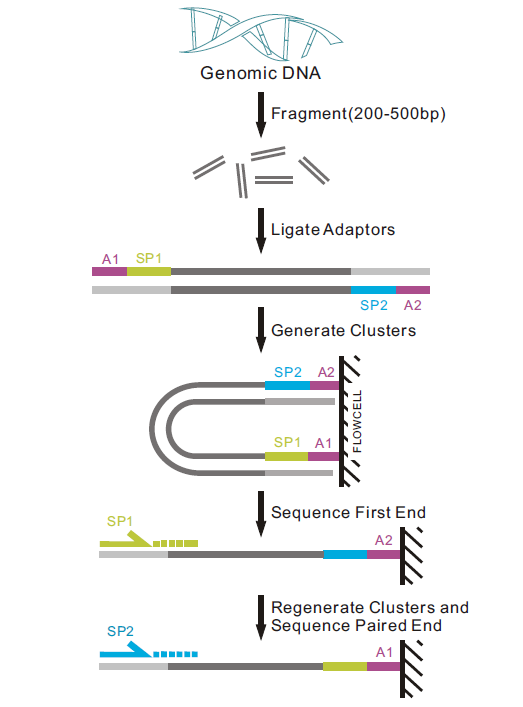
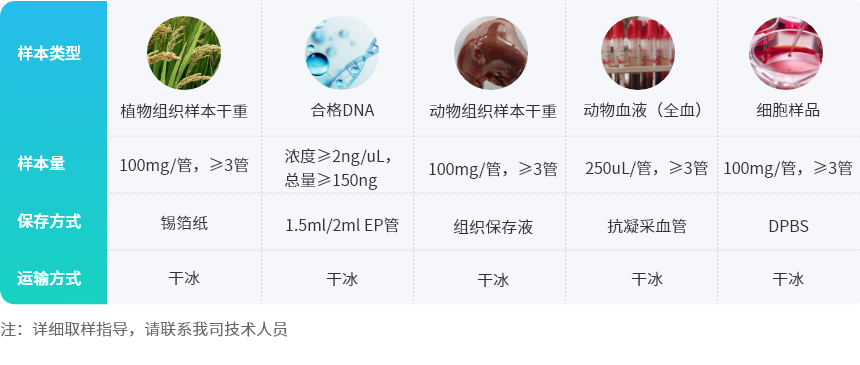
样品基因组DNA检测合格后,利用超声波将DNA序列片段化形成随机片段,对片段化的DNA依次进行末端修复、3′端加A、连接测序接头后,再利用磁珠吸附富集长度为350bp左右的片段,经过PCR扩增形成测序文库。建好的文库先进行文库质检,质检合格的文库通过Illumina平台进行上机测序,测序策略为Illumina PE150,总测序读长为300bp。
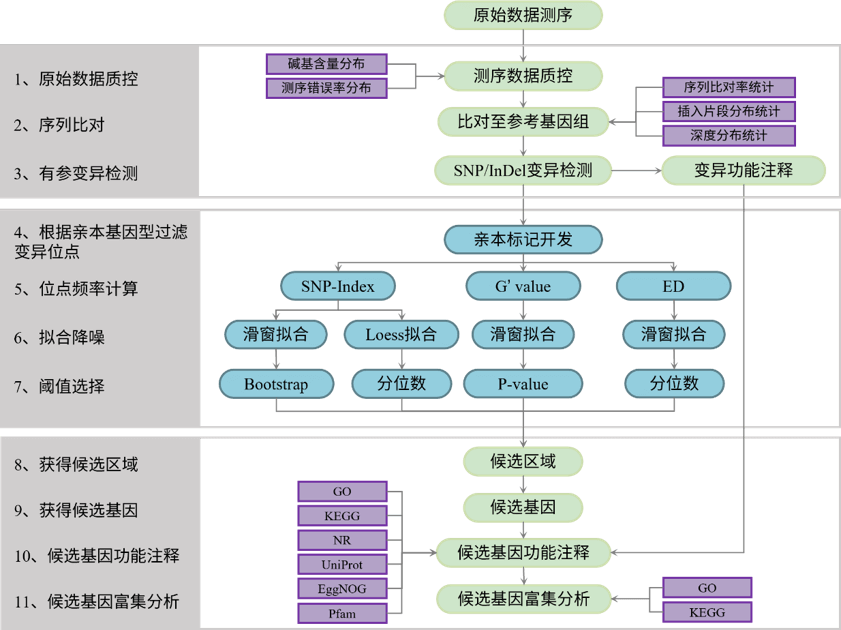
分析流程
在 Illumina测序数据下机之后,对下机数据进行质量控制,过滤其中低质量的数据,获得高质量的数据(Clean Data)。接下来根据GATK官方推荐的Best Practices流程进行变异检测。之后基于SNP和InDel变异位点进行BSA性状定位,后续对候选基因进行功能注释及富集分析
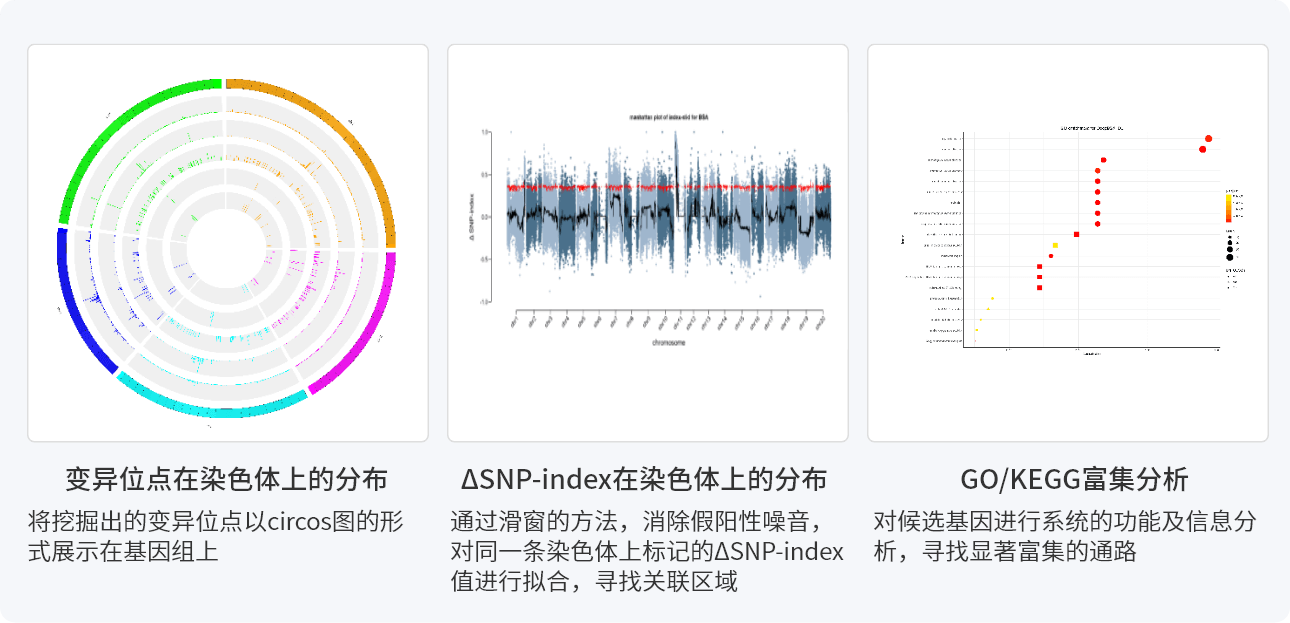
结果展示
应用方向:对关注的目标表型定位候选基因,后续开发分子标记辅助育种,或进一步结合其他组学和分析,挖掘讲述遗传分子机制
SOYBEAN SEED SIZE 1的非同义突变的增强作用在大豆驯化和改良中得到利用
- An enhancing effect attributed to a nonsynonymous mutation in SOYBEAN SEED SIZE 1,a SPINDLY-like gene,is exploited in Soybean domestication and improvement 期刊:New Phytologist 影响因子:10.323 发表时间:2022.09 技术手段:BSA基因定位 研究方向:大豆产量相关基因挖掘 文章链接:doi: 10.1111/nph.18461
研究背景
大豆 (Glycine max) 是从其野生近缘大豆Glycine soja驯化而来的,百粒重是决定大豆产量最重要的驯化性状之一,但人们对其遗传调控基础知之甚少。解析大豆百粒重调控基因及其分子机制,有助于大豆的驯化和改良过程。
技术路线

研究结果
作者使用BSA中的MutMap方法找到了导致种子重量变化的基因GmSSS1,该基因编码SPY同源物,并在控制种子重量和豆荚大小方面具有新的作用。此外该基因第182号位点的突变可以增强GmSSS1的作用,并与大豆的驯化和改良有关。
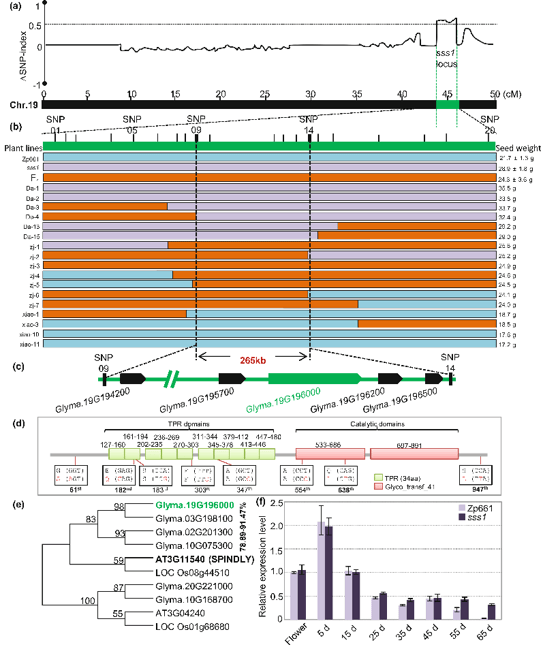

大豆百粒重相关sss1位点定位
常见问题
- 混池的规模多少比较合适呢?
- 通常情况下,混池规模其实越大越好,混池规模是根据群体规模来确定的。美吉推荐老师取群体中最极端的5%-10%的个体(如混池规模至少大于30个样本,即对应的群体规模要大于300个个体才行)。
- 亲本和子代的测序深度推荐多少?
- 通常情况下,测序数据量越大,定位的精度越高。综合考虑,美吉推荐老师亲本单独测序≥10×;子代混池测序平均测序深度≥1×(如50个样本混池,测序深度50x);若基因组或样本量较大,可适当降低测序深度(如100个样本混池,测序深度50×也是没有问题的)。
- BSA缺失亲本可不可以做?
- 可以做。目前主流的BSA分析是2亲本+2混池的模式;但在材料收集过程中可能缺失了亲本或缺失一个子代混池,这种情况也是可以做BSA分析的。但必须有子代混池,1个子代混池时,必须有至少1个相反表型的亲本。










